Niedobór lizosomalnej kwaśnej lipazy (LAL-D) jest rzadkim, postępującym i dziedzicznym schorzeniem spowodowanym mutacjami w genie LIPA, który jest odpowiedzialny za produkcję enzymu, kwaśnej lipazy lizosomalnej (LAL) [1]. LAL-D charakteryzuje się zaburzeniami metabolizmu tłuszczów i cholesterolu w organizmie.
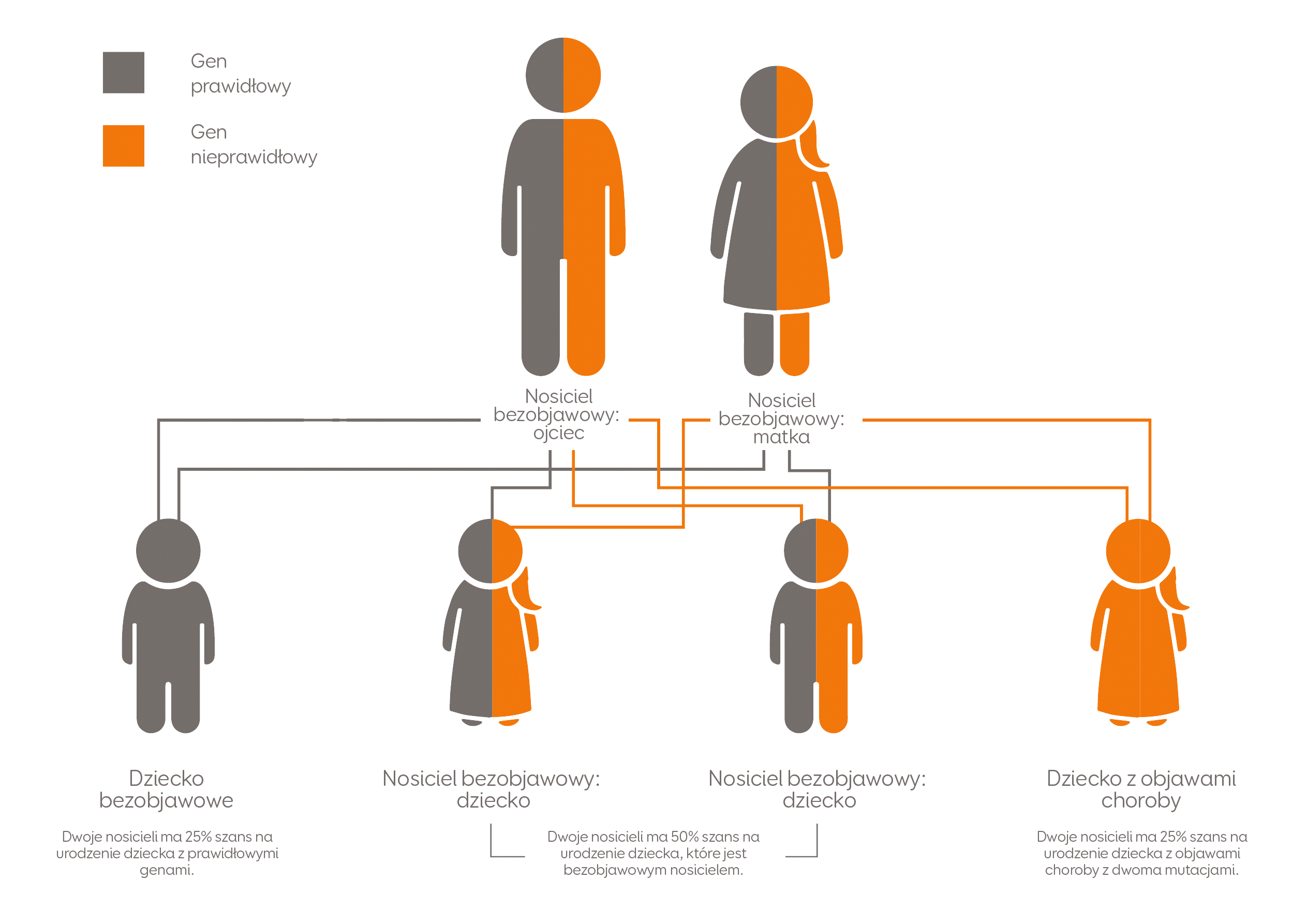
Każda osoba ma dwie kopie genu LIPA. Jedną kopię dziedziczymy po ojcu, drugą po matce. LAL-D występuje wtedy, gdy u danej osoby pojawią się defekty w obu kopiach genu LIPA.
Rys. 1 Schemat dziedziczenia LAL-D. Adaptacja na podstawie materiałów Mayo Foundation for Medical Information and Research. Wszystkie prawa zastrzeżone.
W przypadku rozpoznania LAL-D u dziecka rodzice (będący nosicielami choroby) powinni zostać objęci poradnictwem genetycznym, gdyż ryzyko wystąpienia choroby u kolejnego dziecka wynosi 25% [2].
U pacjentów z LAL-D mutacje w genie LIPA powodują zmniejszoną aktywność LAL lub jej brak, co skutkuje zaburzeniem metabolizmu cholesterolu [1,3].
Piśmiennictwo
Reiner Ž, Guardamagna O, Nair D, et al. Lysosomal acid lipase deficiency — an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014;235(1):21-30.
Mayo Clinic. Autosomal recessive inheritance pattern. Available at: https://www.mayoclinic.org/autosomal-recessive-inheritance-pattern/img-20007457.
Bernstein DL, Hülkova H, Bialer MG, et al. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58(6):1230-1243.
Postępowanie w LAL-D LAL-D można rozpoznać na podstawie wykazania niedoboru aktywności LAL lub potwierdzenia mutacji w genie LIPA[1]. Aktywność enzymu LAL określa się za pomocą enzymatycznego badania krwi ...
Jakie są objawy przedmiotowe i podmiotowe LAL-D The LAL enzyme plays a central role in breaking down certain fats and cholesterol. When there is a decrease or loss of the LAL enzyme, lipids and cholesterol do not get processed and get deposited...
Powikłania LAL-D LAL-D wpływa na wiele ważnych narządów, takich jak wątroba, układ krążenia, śledziona i przewód pokarmowy. W zasadzie większość osób z LAL-D doświadcza powikłań w więcej niż jednym narządzie...