参考資料
1. Reiner Ž, Guardamagna O, Nair D, et al. Lysosomal acid lipase deficiency — an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014;235(1):21-30.
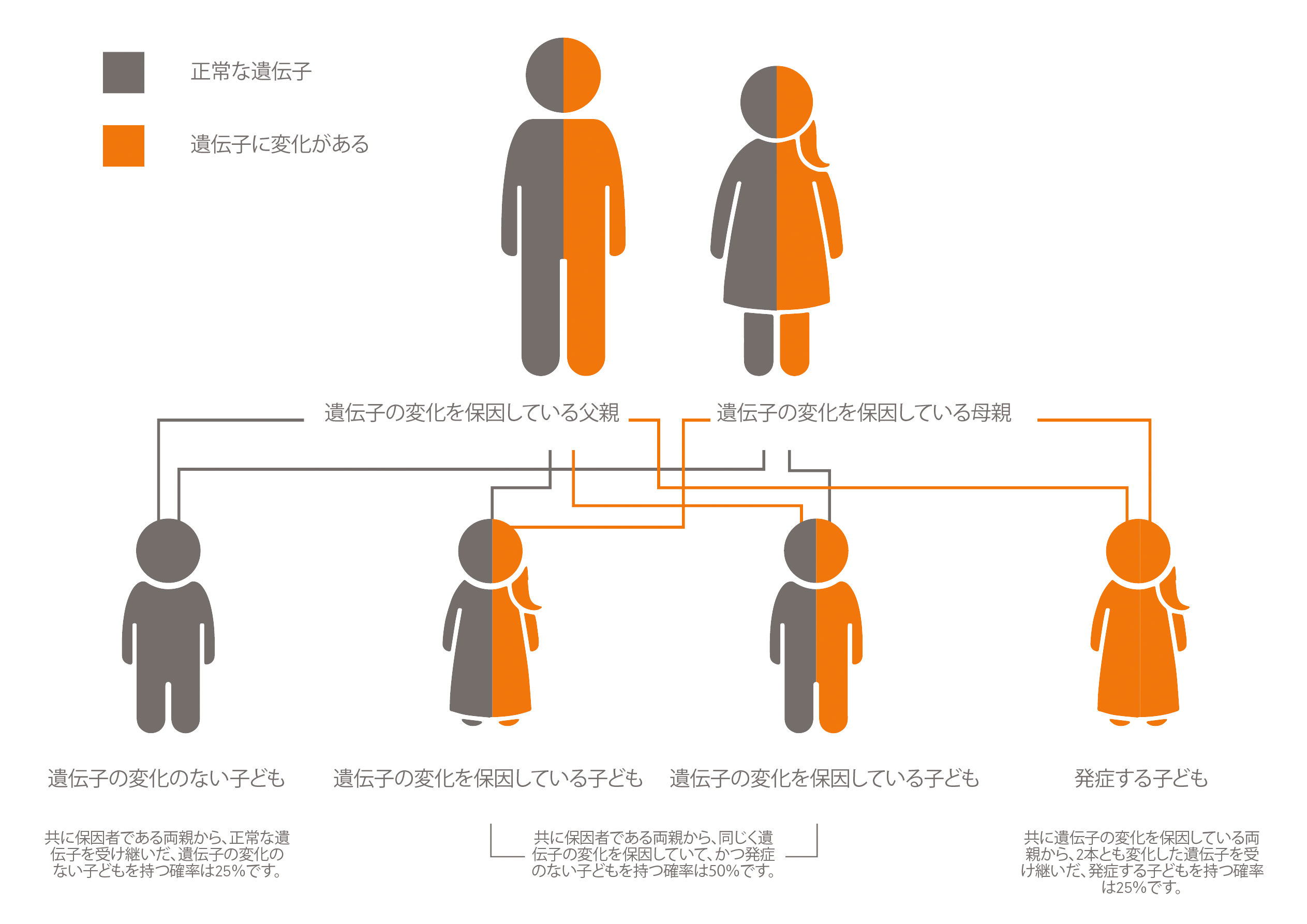
2. Mayo Clinic. Autosomal recessive inheritance pattern. https://www.mayoclinic.org/autosomal-recessive-inheritance-pattern/img-20007457.
3. Bernstein DL, Hülkova H, Bialer MG, et al. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58(6):1230-1243.