Дефицит лизосомной кислой липазы (ДЛКЛ) является редким, прогрессирующим и наследственным заболеванием, вызванным мутациями в гене LIPA, который отвечает за выработку фермента лизосомная кислая липаза (ЛКЛ) [1]. ДЛКЛ характеризуется проблемами с расщеплением и переработкой жиров и холестерина в организме.
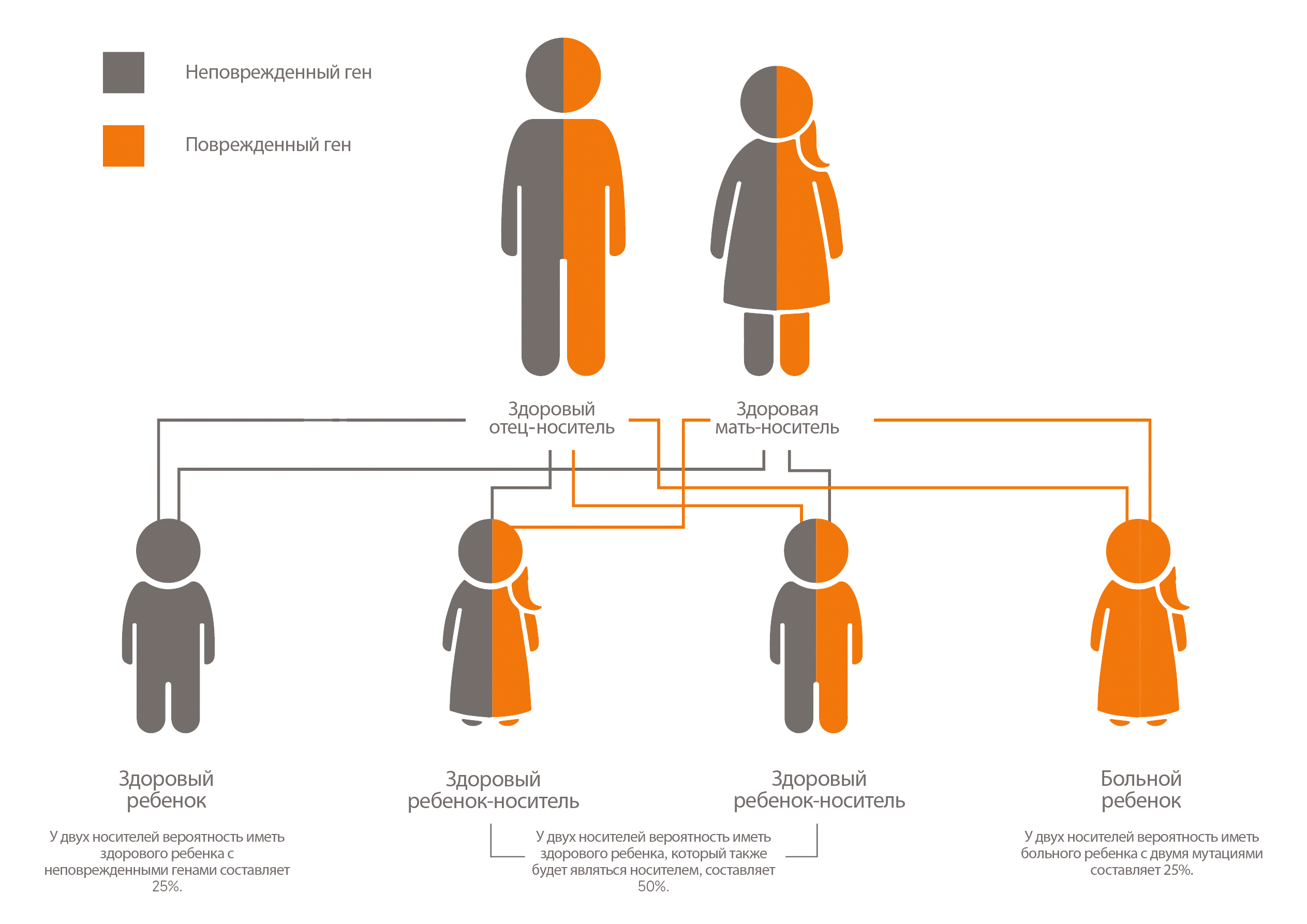
У каждого человека есть две копии гена LIPA. Одна копия наследуется от отца, а другая — от матери. ДЛКЛ возникает, когда у человека есть дефекты в обеих копиях гена LIPA.
Рис. 1 Модель наследования ДЛКЛ. По материалам Фонда медицинской информации и исследований Мэйо. Все права защищены.
Когда у ребенка диагностируют ДЛКЛ, родители (которые являются носителями заболевания) должны пройти генетическую консультацию, так как риск того, что их следующий ребенок будет болен, составляет 25% [2].
У пациентов с ДЛКЛ мутации в гене LIPA вызывают снижение или отсутствие активности ЛКЛ, что приводит к нарушению обмена холестерина [1,3].
Справочные материалы
Reiner Ž, Guardamagna O, Nair D, et al. Lysosomal acid lipase deficiency — an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014;235(1):21-30.
Mayo Clinic. Autosomal recessive inheritance pattern. Available at: https://www.mayoclinic.org/autosomal-recessive-inheritance-pattern/img-20007457.
Bernstein DL, Hülkova H, Bialer MG, et al. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58(6):1230-1243.
Каковы признаки и симптомы ДЛКЛ? Фермент ЛКЛ играет ключевую роль в расщеплении определенных жиров и холестерина. Когда происходит снижение или потеря активности фермента ЛКЛ, липиды и холестерин не перерабатываются и откладываются в различных тканях и органах. Такое накопление вызывает непрерывные повреждения,
Диагностика и лечение ДЛКЛ ДЛКЛ может быть диагностирован путем выявления недостаточной активности ЛКЛ или мутаций в гене LIPA [1]. Уровень активности ЛКЛ определяют с помощью биохимического анализа в сухих пятнах крови [1]. Генетическое тестирование позволяет охарактеризовать генетический статус лиц с подозрением на ДЛКЛ,
Осложнения, связанные с ДЛКЛ ДЛКЛ влияет на многие жизненно важные органы, такие как печень, сердечно-сосудистая система, селезенка и желудочно-кишечный тракт. На самом деле, у большинства людей с ДЛКЛ наблюдаются осложнения более чем в одной системе органов: