Lysosomal acid lipase deficiency (LAL-D) is a rare, progressive and inherited condition caused by mutations in the LIPA gene which is responsible for the production of the lysosomal acid lipase (LAL) enzyme [1]. LAL-D is characterised by problems with the breakdown and use of fats and cholesterol in the body.
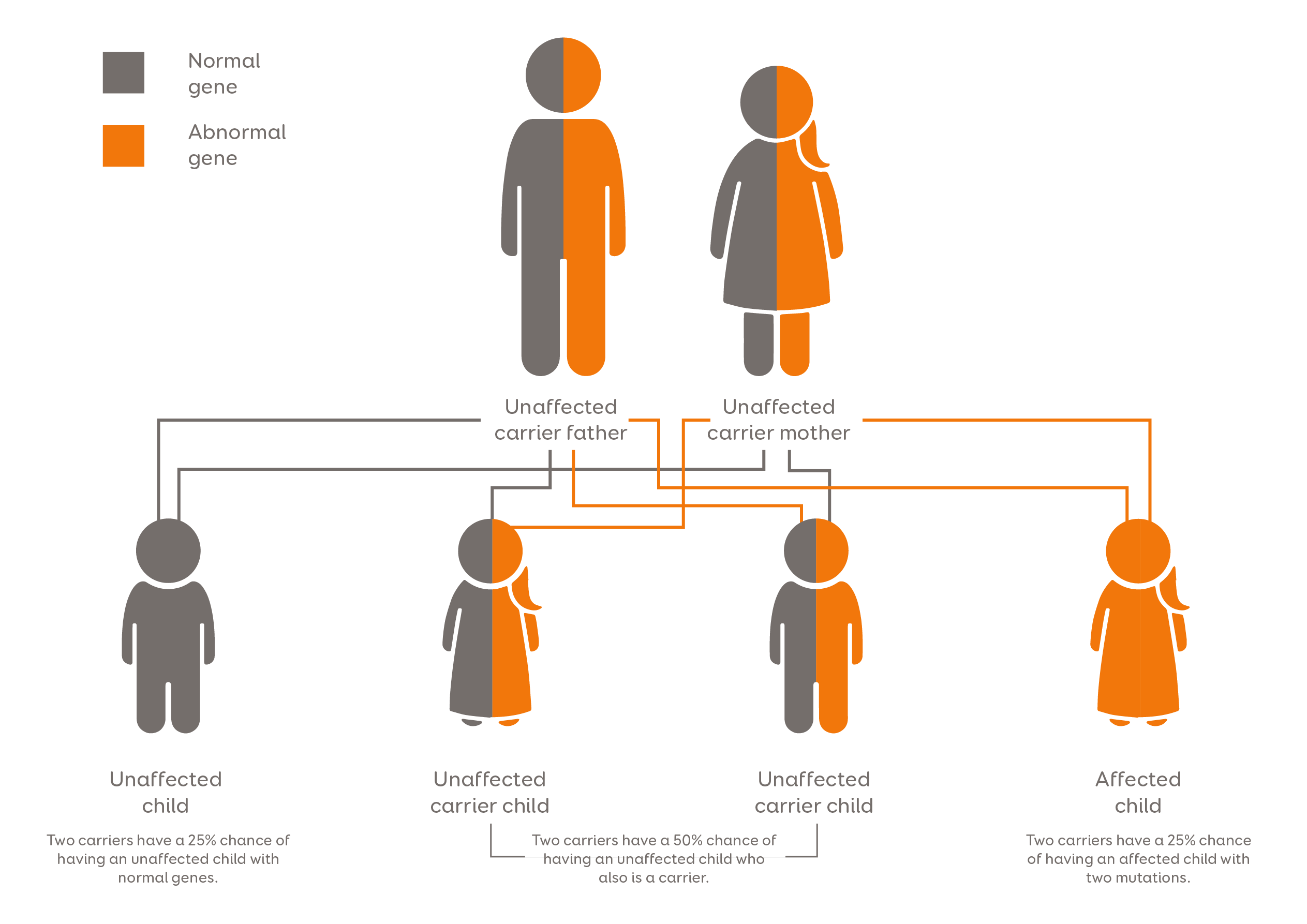
Every person has two copies of the LIPA gene. One copy is inherited from the father and one from the mother. LAL-D occurs when a person has defects in both copies of the LIPA gene.
Fig. 1 LAL-D inheritance pattern. Adapted from Mayo Foundation for Medical Information and Research. All rights reserved.
When a child is diagnosed with LAL-D, the parents (who are carriers of the disease) should undergo genetic testing, as the risk for their next child to be affected is 25% [1].
In patients with LAL-D, mutations in the LIPA gene cause reduced or absent LAL activity, resulting in the disruption of the cholesterol metabolism [1,3].
References
Reiner Ž, Guardamagna O, Nair D, et al. Lysosomal acid lipase deficiency — an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014;235(1):21-30.
Mayo Clinic. Autosomal recessive inheritance pattern. Available at: https://www.mayoclinic.org/autosomal-recessive-inheritance-pattern/img-20007457.
Bernstein DL, Hülkova H, Bialer MG, et al. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58(6):1230-1243.
Complications associated with LAL-D LAL-D affects many vital organs, such as the liver, cardiovascular system, spleen and the gastrointestinal tract. In fact, most people with LAL-D experience complications in more than one organ system: Liver LAL-D is a...
Managing LAL-D LAL-D can be diagnosed by demonstrating deficient LAL activity or mutations in the LIPA gene [1]. The level of LAL-D activity is determined using an enzyme-based blood test [1]. Genetic testing enables characterisation...
What are the signs and symptoms The LAL enzyme plays a central role in breaking down certain fats and cholesterol. When there is a decrease or loss of the LAL enzyme, lipids and cholesterol do not get processed and get deposited...