Il deficit di lipasi acida lisosomiale (LAL-D) è una patologia rara, progressiva ed ereditaria, che è causata da mutazione nel gene LIPA, responsabile della produzione dell’enzima lipasi acida lisosomiale (LAL) [1]. La LAL-D è caratterizzata da problemi a livello di scissione e utilizzo dei lipidi e del colesterolo nell’organismo.
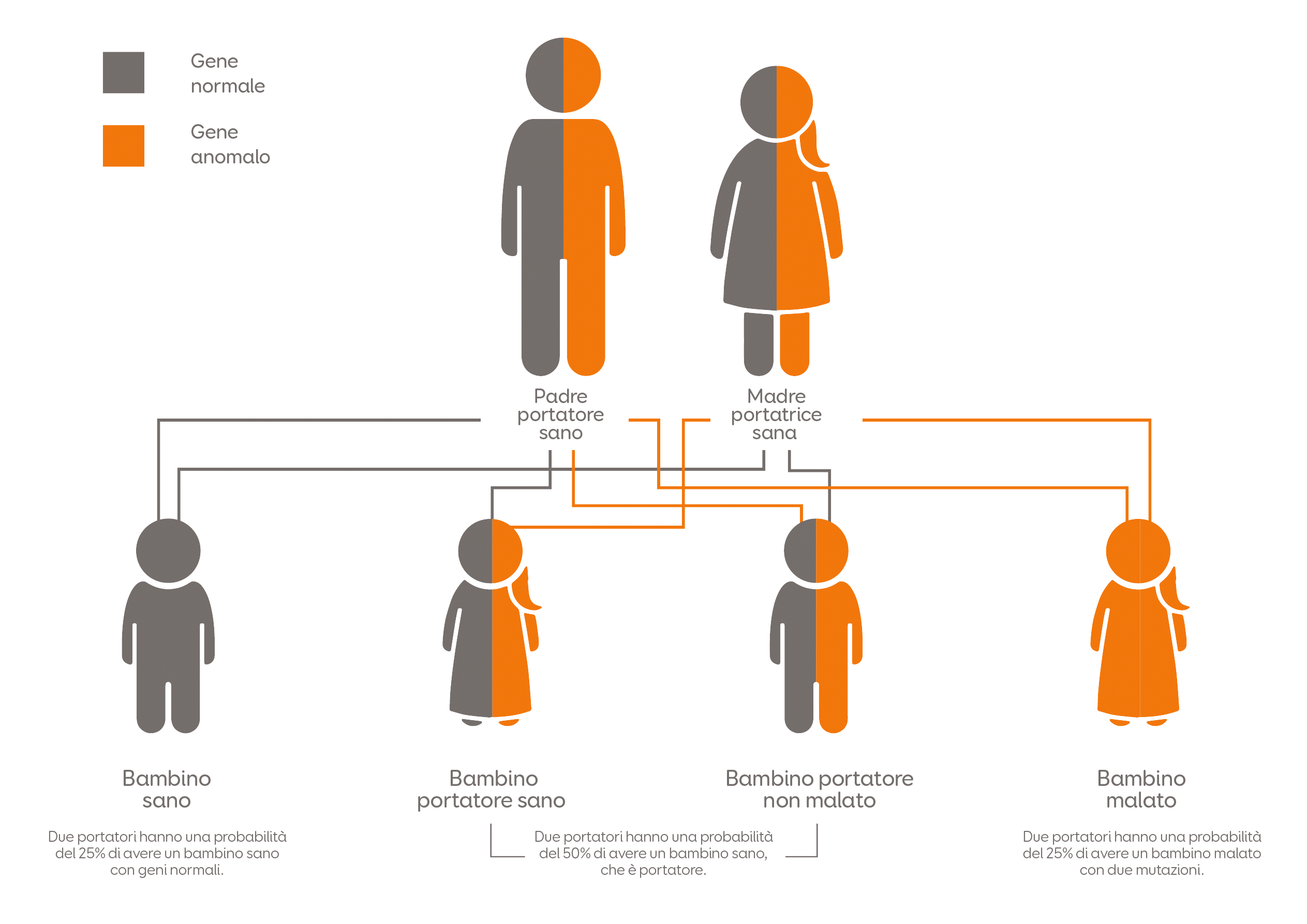
Ogni individuo ha due copie del gene LIPA. Una copia è ereditata dal padre e l’altra dalla madre. La LAL-D insorge quando sono presenti difetti in entrambe le copie del gene LIPA.
Fig. 1 Schema dell’ereditarietà nella LAL-D. Adattamento da Mayo Foundation for Medical Information and Research. Tutti i diritti sono riservati. Quando a un bambino è diagnosticata la LAL-D, i genitori (che sono portatori di questa patologia) devono richiedere una consulenza genetica, perché il rischio di avere un altro figlio con la stessa patologia in futuro è pari al 25% [2]. Nei pazienti con LAL-D, le mutazioni del gene LIPA provocano una riduzione o l’assenza dell’attività della LAL, determinando un’alterazione del metabolismo del colesterolo [1,3].
Referenze
1. Reiner Ž, Guardamagna O, Nair D, et al. Lysosomal acid lipase deficiency — an under-recognized cause of dyslipidaemia and liver dysfunction. 2014;235(1):21-30
2. Mayo Clinic. Autosomal recessive inheritance pattern. Disponibile su: https://www.mayoclinic.org/autosomal-recessive-inheritance-pattern/img-20007457.
3. Bernstein DL, Hülkova H, Bialer MG, et al. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58(6):1230-1243
Complicanze della LAL-D La LAL-D colpisce gli organi vitali, come fegato, apparato cardiovascolare, milza e apparato gastrointestinale. Infatti, molte pazienti presentano anche complicanze in più di un organo...
Quali sono i segni e i sintomi? L’enzima LAL svolge un ruolo centrale nella scomposizione di alcuni grassi e del colesterolo. Quando si verifica un calo o la perdita dell’enzima LAL, non vengono più metabolizzati i lipidi e il colesterolo, che si depositano in vari tessuti e organi...
Gestione della LAL-D La LAL-D può essere diagnosticata, quando si riscontra una carenza di attività dell’enzima LAL o mutazioni del gene LIPA. Il livello di attività dell’enzima LAL può essere valutato tramite l’esame degli enzimi del sangue. I test genetici consentono la caratterizzazione...