Deficit lyzosomální kyselé lipázy (LAL-D) je vzácné, progresivní a dědičné onemocnění způsobené mutacemi v genu LIPA, který je zodpovědný za produkci enzymu lyzosomální kyselé lipázy (LAL) [1]. Onemocnění LAL-D se vyznačuje problémy s odbouráváním a využíváním tuků a cholesterolu v těle.
Každý člověk má dvě kopie genu LIPA. Jedna kopie se dědí po otci a jedna po matce. Onemocnění LAL-D se vyskytuje, pokud má člověk defekty v obou kopiích genu LIPA.
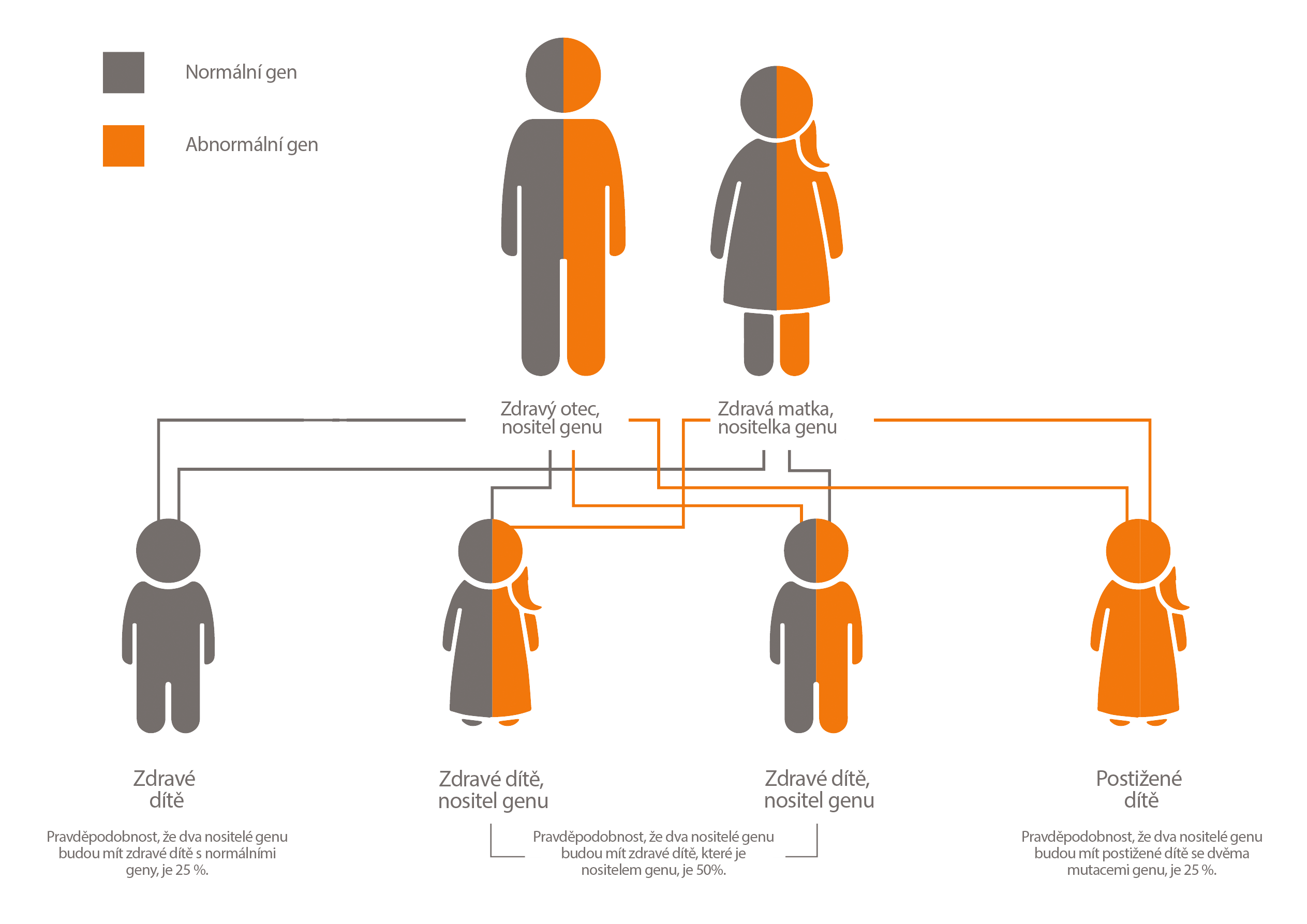
Obr. 1 Vzorec dědičnosti onemocnění LAL-D. Převzato od Nadace Mayo pro lékařské informace a výzkum. Všechna práva vyhrazena.
Je-li u dítěte diagnostikováno onemocnění LAL-D, měli by rodiče (kteří jsou přenašeči onemocnění) podstoupit genetické poradenství, protože riziko postižení jejich dalšího dítěte je 25 % [2].
U pacientů s onemocněním LAL-D způsobují mutace v genu LIPA sníženou nebo chybějící aktivitu LAL, což vede k poruše metabolismu cholesterolu [1,3].
Literatura
Reiner Ž, Guardamagna O, Nair D, et al. Lysosomal acid lipase deficiency — an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014;235(1):21-30.
Mayo Clinic. Autosomal recessive inheritance pattern. Available at: https://www.mayoclinic.org/autosomal-recessive-inheritance-pattern/img-20007457.
Bernstein DL, Hülkova H, Bialer MG, et al. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58(6):1230-1243.
O léčbě LAL-D LAL-D lze diagnostikovat na základě průkazu nedostatečné aktivity LAL nebo mutací v genu LIPA [1]. Hladina aktivity LAL-D se stanovuje pomocí enzymatického krevního testu [1]. Genetické testování umožňuje charakterizovat genetický stav jedinců s podezřením na LAL-D, ale u některých dětí se mohou vyskytovat mutace, které nejsou rutinním screeningem odhaleny...
Jaké jsou známky a příznaky onemocnění LAL-D? Enzym LAL hraje hlavní roli při odbourávání některých tuků a cholesterolu. Při poklesu nebo ztrátě enzymu LAL se lipidy a cholesterol nezpracovávají a ukládají se v různých tkáních a orgánech. Toto skladování způsobuje neustálé poškozování, které může ovlivnit funkci mnoha orgánů v celém těle...
Komplikace spojené s LAL-D LAL-D postihuje mnoho životně důležitých orgánů, jako jsou játra, kardiovaskulární soustava, slezina a gastrointestinální trakt. U většiny lidí s LAL-D se vyskytují komplikace ve více než jednom orgánovém systému...